
Updated September 07, 2022
Implementation of the FDA’s long-awaited unique device identification (UDI) system for medical devices is now a must, and the requirements for manufacturers are significant. Device makers must utilize an efficient process to generate the large volume of high-quality data they’ll need for compliance.
A UDI is a two-part numeric or alphanumeric code used to identify a medical device. It has a mandatory, fixed portion called the device identifier (DI), and a conditional, variable portion called the production identifier (PI). The DI shows the labeler of the device (this is usually the manufacturer) and the specific version or model. The PI holds production control data such as:
Congress in 2007 passed legislation that required the FDA to establish a UDI system for all regulated medical devices, from bandages to artificial heart valves. The legislation was intended to improve patient safety by enabling the FDA, manufacturers and healthcare providers to more quickly identify devices and correct problems. In September 2013, the FDA released its long-awaited final rule on the system, which set UDI requirements in three main areas as outlined below. The FDA further codified this guidance in the 2020 UDI Compliance Policy Guidance:
Labeling – Labelers must obtain a UDI from a federally accredited agency and include it on the label and packaging of all devices, in both plain text and a machine-readable format, such as a bar code or radio-frequency identifier. Any dates on the label must use the format YYYY-MM-DD.
Direct marking – Multi-use devices intended to be reprocessed (cleaned, sterilized, etc.) before reuse must be directly marked with a permanent UDI.
Data submission – For all devices, the labeler must submit detailed device information to the FDA’s global unique device identification database (GUDID) and update it as needed. The GUDID is publicly searchable, so end users can look up product information, verify that a device isn’t a counterfeit, check on recalls, etc.
Industry feedback on an earlier proposed rule led to a number of revisions in the final version. One of the most significant is that implantable devices do not have to be directly marked. Another change is that single-use products, such as bandages, are exempt from compliance if they are distributed and stored until use in a larger package. The individual parts of convenience kits and combination products also do not need UDIs, although the kits and combination products do.
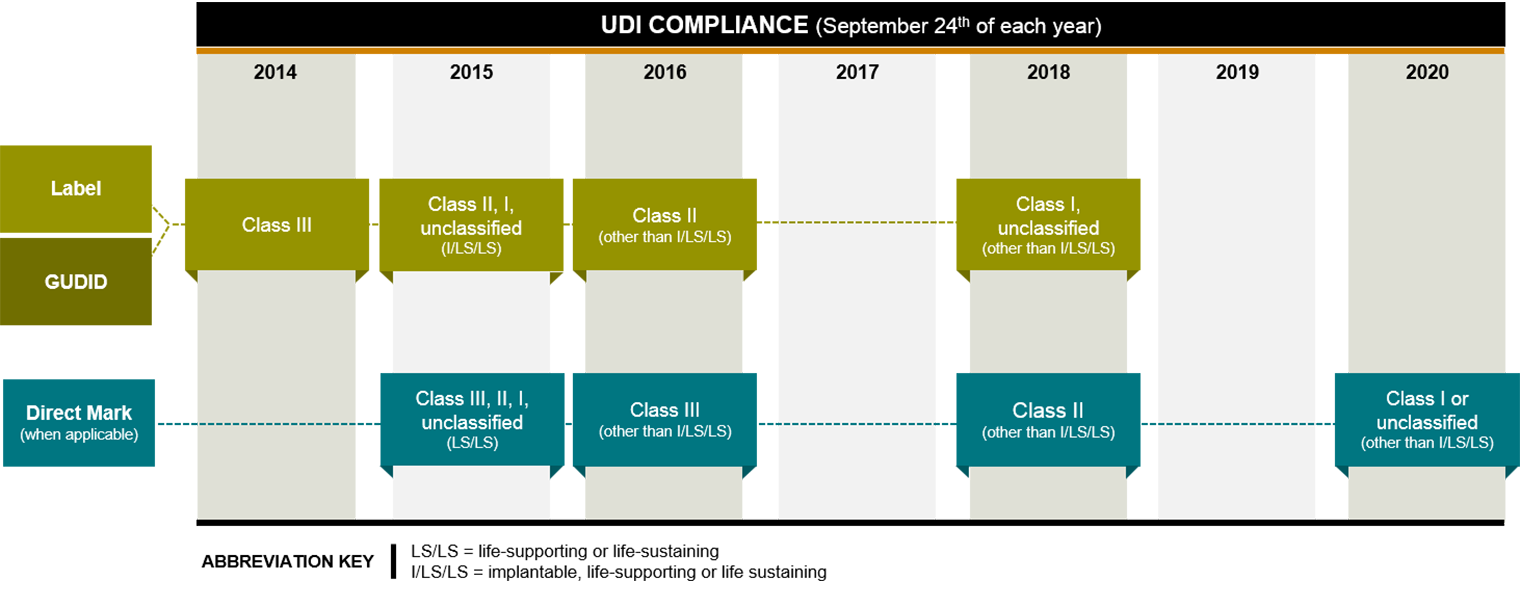
Different classes and types of devices were given different compliance deadlines, with lower risk devices―which are the majority―having more time to comply. First up were high-risk Class III devices, such as pacemakers, which generally had to meet a September 2014 deadline for labeling and GUDID submission. Only about 10% of medical devices are Class III, according to the FDA.
As of September 24, 2015, Class II, I and unclassified implantable, life-sustaining and life-supporting devices (tracheal tubes, for example) must meet the labelling and GUDID requirements, and all multiuse life-supporting and life-sustaining devices that are meant to be reprocessed must meet the direct marking requirement.
The labeling and GUDID submission requirements were phased in for all devices by September 2018, and the direct marking requirement was fully phased in by September 2020. For existing inventory that was already labeled, the final rule allows a grace period of three years after the device’s compliance deadline. Manufacturers will have to re-label any remaining inventory when the grace period ends this time next year, 2023.
In the FDA’s updated guidance released on July 25, 2022, the authority acknowledges that new policy for consumer health products is forthcoming in September of this year. In order to give consideration to companies who must adhere to the policy, enforcement of GUDID submission requirements will begin December 8, 2022. The FDA stated, “We believe this brief extension of the policy may help facilitate submission of high quality UDI data to GUDID and is consistent with the public health.”
Companies that miss their deadline are disallowed from selling their device across state lines in the U.S. They also risk damaging their reputation.
To create a compliance process, device makers must first figure out the overall scope of UDI requirements for all their devices. This may seem simple, but it can be a time-consuming task for manufacturers with a multitude of products or product variations.
Device companies also need to determine who the labeler is, and who is responsible for what data. Externally, this can mean working with contract manufacturers to clearly define which entity handles UDI-related tasks. Internally, it means figuring out which department will gather specific information, how it will be stored, who will submit it to the GUDID, and who will manage it on an ongoing basis.
The data requirements are extensive: For each UDI, labelers need to compile approximately 60 different data points. They must create labels, and they must submit device information to the GUDID in a compliant format. Submissions can be done manually using the FDA’s online interface―a time-consuming and error-prone process―or by a more efficient bulk upload, using an HL7 SPL-compliant XML file. Whenever there is a product change, such as a device upgrade, the labeler must make any needed label changes and resubmit information to the database.
Data quality is critical. Submitters have only seven days to correct mistakes, and after that, the FDA will label incorrect submissions as noncompliant. It’s also important to note that GUDID submission is continual, not periodic. This means the process must be repeatable, and the data must be reliable.
Using ERP-generated information will save manufacturers from having to manually “scrub” data to ensure that it’s correct. Device makers can use their ERP system to ensure data accuracy and streamline labeling, direct marking and GUDID submission. (This may require some integration work with other systems, or a master data management system.)
To simplify data submission, an ERP package can be customized to restructure device information into the HL7 SPL format and upload it to the GUDID. Another option is to run a report in ERP and then either outsource the HL7 SPL formatting/submission or use a subscription service to do it.
An ERP system can natively generate device data in a label-ready format, which can be printed on a physical label using a third-party tool. The system can also be integrated with shop floor equipment, to allow manufacturers to call up accurate UDI information when they are printing a permanent mark directly onto a device. With the advancement of new technologies—Microsoft Power Platform, UIPath, and many others—supporting business process automation, Armanino sees clients embracing their own automated business-specific app to manage GUDID information and upload to the FDA.
The FDA has said from the beginning that the system is meant to evolve. An efficient UDI process will likely become even more important for manufacturers as UDI data finds new uses and users.
For device makers, UDIs could provide a way to generate valuable post-market performance data. They could use this information, for example, when requesting FDA approval of a new use for a device.
Adoption of UDIs by healthcare providers―which is needed to truly improve patient safety―will be a critical variable in the next few years. Although many providers have had to make costly updates to their clinical and operational information systems in order to use UDIs, most recognize the potential safety and supply chain efficiency benefits. UDI data also helps providers track device effectiveness―which is increasingly important as the healthcare industry continues shifts from a focus on volume to a focus on quality of care.
Providers do not have to use UDIs, with one main exception: The FDA now requires facilities such as hospitals, nursing homes and ambulatory surgery centers to include UDI information when they report on patient deaths involving a Class III device. The Department of Health and Human Services also may require providers to report UDI data on implantable devices in the upcoming stage 3 of its meaningful use incentive program, which promotes the use of electronic health records for Medicare and Medicaid patients. In addition, some healthcare industry groups want to see UDI information incorporated into claims data.
UDI implementation in the U.S. will also help pave the way for a global system. Although the FDA is the furthest along, regulators in the European Union and countries such as Brazil, Canada, Australia, China and Japan are creating their own UDI guidelines for medical devices and working with the FDA to build a global regulatory framework.
Compliance requires a lot of heavy lifting by device makers, and there will no doubt be more shifts in what data is required, who uses it and how. With rollout of the system fully deployed for all devices by end of 2022, manufacturers need to assess their own level of UDI readiness, including their ERP and other systems, and start working toward meeting their requirements. The latitude given to the industry over the past 5 years is finally at its end. It is imperative that businesses move responsibly and swiftly to build an efficient and compliant UDI process now. Requirements will undoubtedly continue to evolve, but the need for device makers to manage and provide high quality data will only increase.
Contact our life sciences industry experts to discuss your compliance process.



